AmpliSAS - Amplicon Sequencing ASsignment tool
AmpliSAS
accomplishes a full analysis of the data: de-multiplexation, clustering and filtering of variants (unique sequences) with genotyping purposes.
After running AmpliCHECK
we should be able to establish the length of the desired PCR products (markers),
the % of errors in variants and a threshold frequency to decide if a variant is real or is an
artefact. Then we can run AmpliSAS to perform an exhaustive analysis and genotyping.
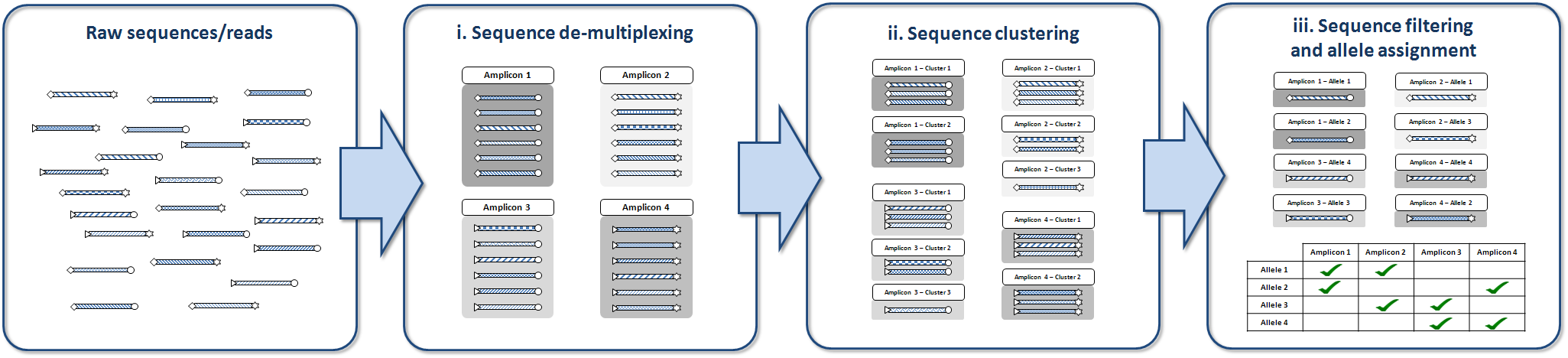
AmpliSAS workflow is divided into three main steps:
- De-multiplexing of reads into amplicons and unique sequences based on matching of primers and tags sequences.
- Clustering of amplicon sequences, where potential alleles and artefacts are grouped together based on user-defined thresholds.
- Filtering of sequences based on user-defined parameters, like number of samples, frequency, depth, chimeras and frameshifts detection...
AmpliSAS takes as input:
- SEQUENCE FILE: FASTQ or FASTA format file (compressed or uncompressed). Multiple sample/amplicon sequence files should be packed into a unique .ZIP or .TAR.GZ file. Also previously analyzed results can be used as input in AmpliSAS format Excel file.
- AMPLICON DATA 1: primer and tag information in a CSV format file as explained in the documentation.
Results can be downloaded on the same page or from an email message after analysis completion.
For more information, read the documentation.
Run AmpliSAS
Disclaimer
Your use of any of these tools is at your own risk. We do not give any representation or warranty nor assume any liability or responsibility for the data nor the results posted (whether as to their accuracy, completeness, quality or otherwise). Access to these data is available free of charge for ordinary use in the course of research. By visiting the site, you accept our use of cookies and you accept that your data and results will be stored in our server.